
2. Proteins#
2.1. Amino acids#
2.1.1. Monomers and polymers, amino acids and proteins#
Just like nucleobases are the monomeric units of nucleic acids, monosaccharides are monomers of polysaccharides, amino acids are the monomeric building blocks of peptides (also called polypeptides) and proteins.
NEED TO KNOW
When present in a polypeptidic sequence, amino acids are called residues!
As their name suggest, the defining structural features of amino acids are a central carbon, the alpha carbon or C\(\alpha\), bound to a primary amine and a carboxyl acid. The alpha carbon finds itself at the center of a tetrahedron: aside the primary amine, and the carboxyl group, and a hydrogen atom, what distinguishes each amino acid is the group connected to the C\(\alpha\), called the R group or side chain.
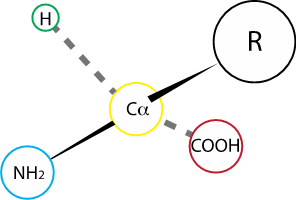
Fig. 2.1 Amino acids consist in 4 different chemical groups arranged in space. Dotted lines indicate bonds that extend behind the plane. Solid lines represent bonds that extend towards the reader.#
GOOD TO KNOW
Because of their three-dimensional arrangement, amino acids are chiral. That is, they exist in two different isoforms, stereoisomers, which are chemically identical, but their spatial orientation differs.
2.1.2. Amino acids are charged objects#
Many properties of amino acids and peptides that are useful for biochemists to know are based on pH. In solution, at neutral pH, amino acids exist as zwitterions.
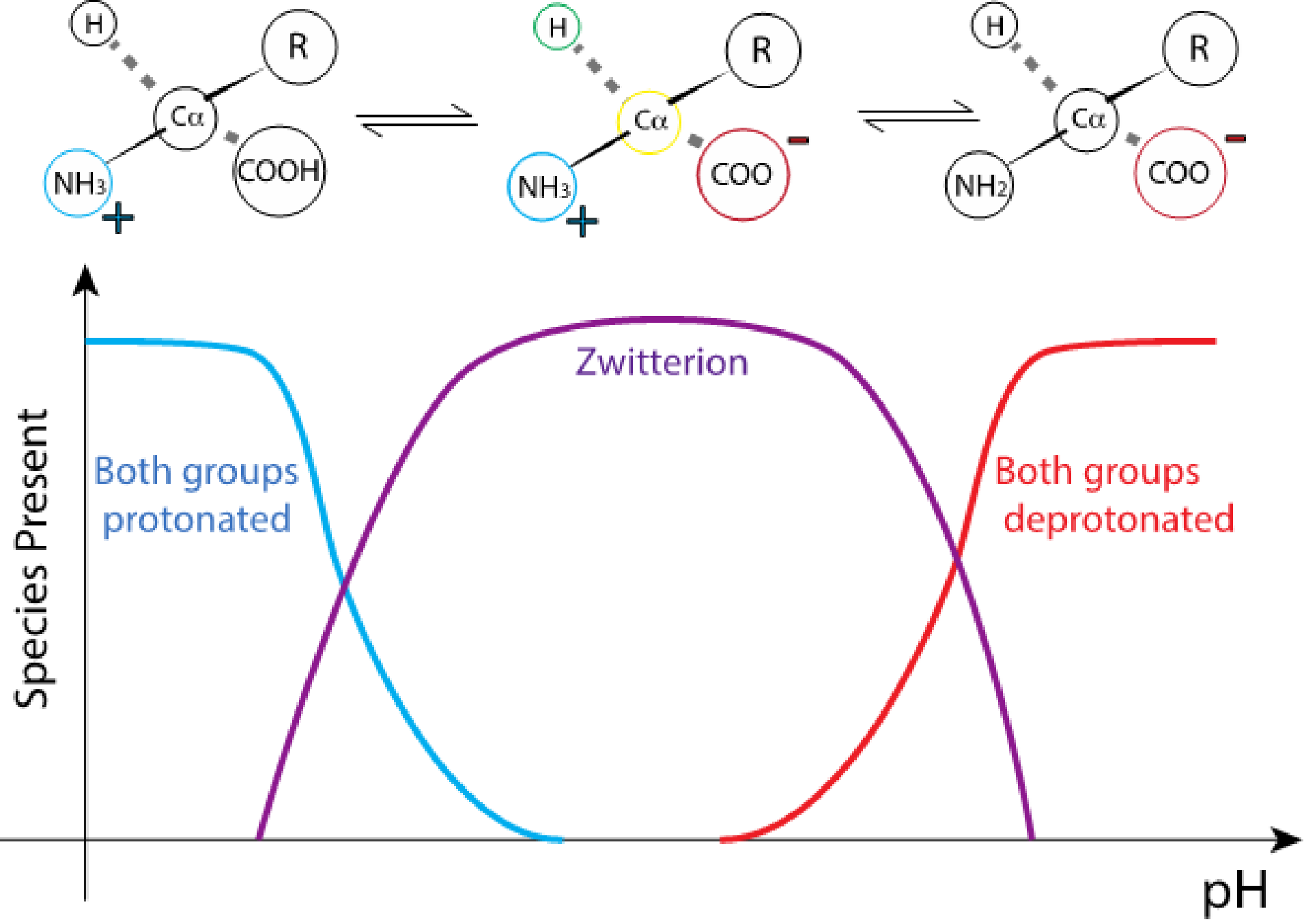
Fig. 2.2 The net charge of an amino acid depends on the pH of the solution.#
The protonation state of the amine and the carboxyl group is dependent on the pH of the solution. The carboxyl group will be deprotonated for pH values higher than ~2, while it takes a pH higher than about 9 to deprotonate the amine of an average amino acid. Therefore, at neutral pH (~7-7.6), the net charge of an amino acid will be largely determined by its R group. Depending on the chemical nature of the side chain, there will be one pH value where each amino acid will be found at neutral net charge.
But how does this property of amino acids affect proteins?
Each protein is characterized by its isoelectric point (pI), the pH value in solution where they carry no electric charge, due to the protonation state of its chemical groups. The isoelectric point depends on the primary sequence of the protein, and is therefore unique to each.
GOOD TO KNOW
The isoelectric point of a protein is a useful property to keep in mind: it is also the pH value at which the protein is least soluble in water!
2.1.3. Amino acid Structures#
In natural amino acids, there are 20 different R groups.
GOOD TO KNOW
In higher living organisms, proteins include 20 different L-aminoacids, which are called natural amino acids. Natural amino acids are the only ones recognized and utilized by the cellular machinery for protein translation. Yet, from the chemical definition of amino acids, many non-natural amino acids exist! They are not proteinogenic, that is, they are not included in proteins synthesized in nature, either because they are D-stereoisomers, or because no loading t-RNA exist for them - their side chain is not among the 20 natural ones. Some of them can be found in nature, some are biochemically synthesized for genetic engineering or other commercial application, you will learn about this in other courses.
The R groups can be roughly divided in several categories according to their physical properties:
2.1.3.1. Non-polar, Hydrophobic side chains#
Fig. 2.3 Alanine.#
Fig. 2.4 Valine.#
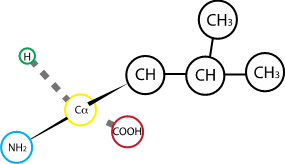
Fig. 2.5 Leucine.#
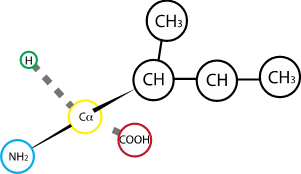
Fig. 2.6 Isoleucine#
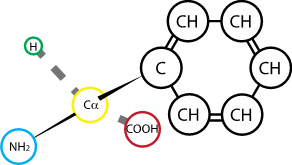
Fig. 2.7 Phenylalanine#
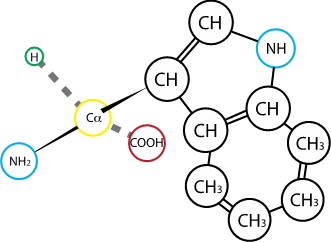
Fig. 2.8 Tryptophan#
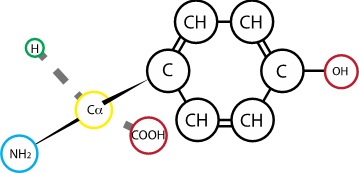
Fig. 2.9 Tyrosine#
2.1.3.2. Uncharged, polar side chains#
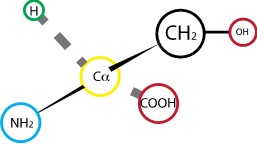
Fig. 2.10 Serine.#
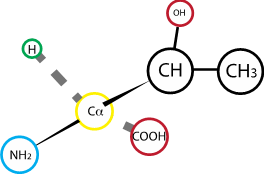
Fig. 2.11 Threonine#
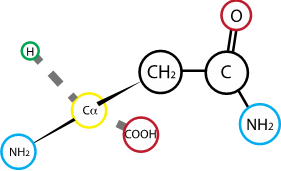
Fig. 2.12 Asparagine#
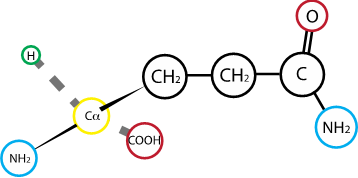
Fig. 2.13 Glutamine#
2.1.3.3. Positively charged side chains#
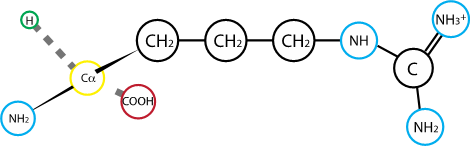
Fig. 2.14 Arginine. R group pKa: 12.10#
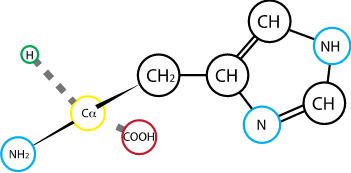
Fig. 2.15 Histidine. R group pka: 6.04#
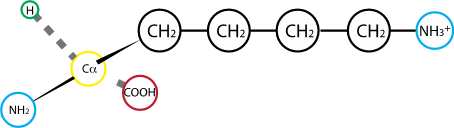
Fig. 2.16 Lysine. R group pKa: 10.67#
2.1.3.4. Negatively Charged side chains#
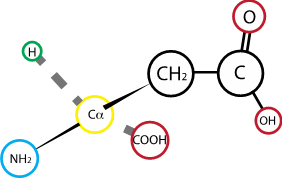
Fig. 2.17 Aspartic acid (aspartate)#
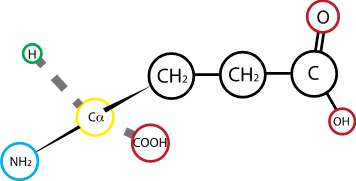
Fig. 2.18 Glutamic acid (glutamate)#
2.1.3.5. Special amino acids#
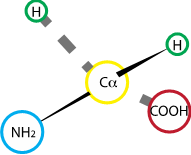
Fig. 2.19 Glycine#
Glycine features a hydrogen as an R group. As the smallest R group, glycine is also the most flexible residue within a protein sequence, and is often found in hinges and/or unstructured domains.
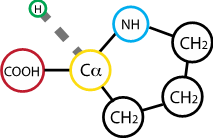
Fig. 2.20 Proline#
Proline is the only natural amino acid whether the alpha carbon is found in a ring within the R group. As such, the peptide bond it will form with neighboring residues cannot rotate with the same degree of freedom of most others. Proline is therefore the most sterically constrained among amino acids; its presence imposes a bend on the secondary sequence of the protein.
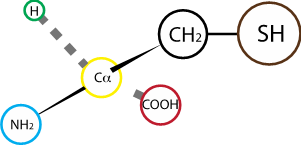
Fig. 2.21 Cysteine. R group pKa: 8.14#
Cysteine contains a thiol reactive group on its side chain. Depending on the oxidoredox state of their environment, cysteines can form disulfide bonds with other cysteine residues. Disulfide bonds have an important role in protein folding and stability.
GOOD TO KNOW
Most cytosolic compartments are reducing environments, where cysteines prefer to remain in their reduced – unbound – state. Therefore, disulfide bonds are mostly found in proteins that are meant to be secreted.
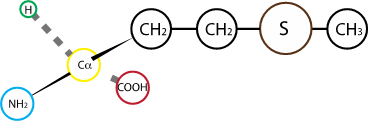
Fig. 2.22 Methionine.#
Methionine is the amino acid encoded by the START codon, and as such, is found as the first residues – N terminal residue - in most protein sequences. Its side chain is hydrophobic, but is different from the bulk of amino acids with hydrophobic side chains as it includes a sulfur atom.
2.1.3.6. Selenocysteine and pyrrolysine#
If you have already counted 20 amino acids so far, and you wonder why there is yet another paragraph to cover: congrats, you passed the GorillaTest of this Biochemistry course (if you are curious to learn what a gorilla test is, you can look here after you complete this chapter)!
In addition to the 20 amino acids described here, 2 additional, less common residues have been identified: selenocysteine and pyrrolysine.
Selenocysteine shares the structure of cysteine, where a selenium atom (Se) replaces sulphur (S). It can be found in higher eukaryotes, including humans. Pyrrolysine, discovered in 2002 [1], has only been found in protein sequences of specific archaeal organisms so far.
2.2. The peptide bond#
Amino acids are the monomers that bind to form peptides and proteins. The chemical bond(s) that link one residue to another in a polypeptide is always of the same type: a peptide bond, a covalent bond between the nitrogen of the primary amine of one amino acid (that becomes a secondary amine in the peptide!) and the carbon of the carboxyl group in the next residue.
The peptide resulting from this condensation reaction has an N terminal and a C terminal.
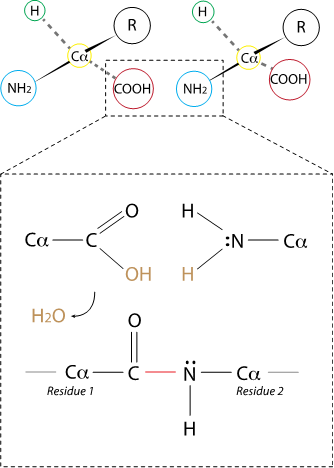
Fig. 2.23 The peptide bond connects two adjacent residues in a peptide chain via the carboxyl group of one residue and the amine group in the next one. In the cell, the condensation reaction is catalyzed by ribosomes, and produces the peptide and one molecule of water.#
Short aminoacidic chains are polypeptides, or peptides; above the threshold of 30-50 residues, the polypeptide chain is usually called a protein. There is no definite threshold length that distinguishes a long peptide from a short protein.
2.3. The Ramachandran Plot#
The properties that make proteins different between each other, and the central acting molecules in the cells, stem from the physical properties of the amino acids and the peptide bond.
The property that makes the peptide bond special is the spatial arrangement of the atoms that form the bond itself. Although the nitrogen and the carbon share one couple electrons, making the peptide bond a single covalent bond, the electrons of the C=O bond are partially shared with the electrons of the peptide bond. This nature of the peptide bond has two important consequences:
It gives the peptide bond a partial status of double bond, and
It positions the atoms forming the bond on a plane, making it impossible for the bond to freely rotate.
The only rotation possible are the rotations of the bonds of the \(\alpha\) carbon \(\phi\) and \(\psi\), which has crucial repercussions on the conformations that a protein is allowed to adopt!
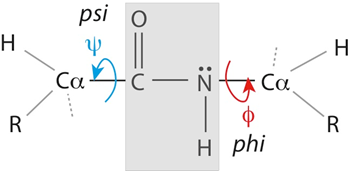
Fig. 2.24 The chemical bonds adjacent to the peptide bond have a degree of freedom to rotate (red and blue), while the peptide bond itself is found on a rigid plane (grey).#
The bonds in a peptide chain cannot assume all the possible \(\phi\) and \(\psi\) angles: the atoms would clash because of steric hindrance. Only angles that position the atoms in way to not produce clashes are permitted. The permitted angles can be reported in a Ramachandran plot as the one in Fig. 2.25.
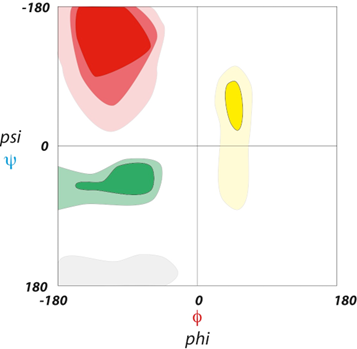
Fig. 2.25 The bonds in a peptide chain cannot assume all the possible \(\phi\) and \(\psi\) angles: the atoms would clash because of steric hindrance. In a virtual space graph, not all regions can be occupied. The accessible regions of space, which are colored in the figure, define structures with specific \(\phi\) and \(\psi\) angles: beta sheets (red), alpha helices (green), and left-handed/collagen alpha helices (yellow).#
GOOD TO KNOW
Some combination of \(\phi\) and \(\psi\) angles are more favorable, and for this reason they tend to be found more often in proteins. The angles that residues assume in regard with each other allow them to assume specific conformations, that falls into a few categories to form motives.
2.4. Protein structure#
When we look at proteins, their conformation – the 3D structure they assume in the cell - is as important as their aminoacidic sequence. Most proteins cannot perform their function if their aminoacidic sequence organizes in space as a simple string of residues. The structural arrangement of protein in space is so important, that some cellular proteins, chaperones, are devoted to ensure correct folding of other proteins!
NEED TO KNOW
The structure of a protein is tightly connected to its function.
How do protein sequences arrange in three-dimensional space?
By combining 20 different amino acids, the possible theoretical sequences for a protein composed of \(n\) residues are equal to
One could expect that the number of existing protein structures is potentially as big as the number of protein sequences – each protein folds into its own shape.
In reality, this does not happen. Thousands of proteins exist, but they tend to assume a finite number of conformations. Different hierarchical levels of possible protein structure exist.
NEED TO KNOW
The structural and physical properties of proteins are ultimately determined by their primary structure. The sequence of residues, each with their own side chains, in a protein results in both unique structures – because of the permitted and preferred combinations of psi and phi angles of each residue – and properties – because of the nature and special alignment of different side chains!
2.4.1. Primary structure#
The primary structure of a protein (or a peptide) is the first level of organization of the chain, and consists in the aminoacidic sequence that compose it – from the N terminal residue, to the C terminal residue.
2.4.2. Secondary structure#
The secondary structure is defined as the reciprocal 3D arrangement in space of short aminoacidic sequences linked to each other by peptide bonds within a protein. Secondary structures arrangements are therefore limited by the possible phi and psi angles, described by the Ramachandran plot (described in The peptide bond), which depend on both the single amino acid in each position, and the combination of neighboring residues within that region of the protein.
Unlike the primary structure, held together by covalent bonds, different types of non-covalent bonds can contribute to secondary structures.
Hydrogen bond
Electrostatic bond
Disulfide bond
Two main secondary structures exist: the alpha-helix (\(\alpha\) helix), the beta sheet (\(\beta\) sheet).
2.4.2.1. The alpha-helix (\(\alpha\) helix)#
Alpha helices are so called, because the backbones of the residues that form this type of secondary structure are arranged in a right-handed helix. The helix is stabilized in a regular shape and pace by hydrogen bonds that form between the carboxyl group of each residue R and the amine group of the residue R+4. For this reason, \(\alpha\) helices are characterized by a regular number of residue per turn (3.6) and a regular helix pitch (0.54 nm), no matter in which protein they are found.
2.4.2.2. The beta sheet (\(\beta\) sheet)#
Beta sheets are planar structures in which neighboring amino acids assume a regular planar arrangement and are extended in respect to each other, rather than being brought together by hydrogen bonds, like the alpha helices are. In this type of secondary structure, the hydrogen bonds form between adjacent short portions of extended sequences; all hydrogen bonds are found on the same plane, hence the nomenclature sheet.
Beta sheets are antiparallel if the hydrogen bonds form in between chains oriented in the opposite direction (N-to-C and C-to-N), or parallel if the hydrogen bonds form between chains oriented in the same direction (N-to-C and N-to-C). They can also be formed by multiple combinations of sheets arranged in mixed orientations.
2.4.2.3. Other secondary structures#
Among secondary structures quantitatively less represented in proteins are different helices, the beta turn, and the random coil, a seemingly less defined structure that is nevertheless important in intrinsically disordered proteins (IDPs).
2.4.3. Tertiary structure#
Secondary structures exclusively define three-dimensional arrangements in space of aminoacidic chains. Per se, secondary structures are not associated with specific protein functions. In proteins, secondary structure elements combine in tertiary structures, the next higher hierarchical level of arrangement.
A motif is a short sequence pattern that folds in similar ways, and can be associated with a specific function. They are characterized as tertiary structure because, unlike described for secondary structure elements, they can, and usually are, bring lose together residues that are far away from each other in the primary sequence.
Motives can have a structural function (zinc-fingers, loops, helix bundles…), other form a binding platform for other macromolecules (helix-turn-helix, beta-barrel).
NEED TO KNOW
Since the possible protein structural conformations are finite, two aminoacidic chains can fold in the same motif even if they have different sequences!
Longer protein regions that perform a specific function, or that share a structural conformation within the protein, are defined as domains. Domains are much more numerous than motives and, unlike motives, are usually found in a wider variety of structural conformations. The overall arrangement of domains within a protein, and in general the overall structural shape of a protein, goes under the name of tertiary structure.
2.4.4. Quaternary structure#
Proteins described so far perform a function thanks to the structure that their primary sequence folds in. It would seem therefore as any cellular function can be performed by a single aminoacidic chain, provided that it is folded correctly in its tertiary structure. In reality, some of these functions are performed by proteins that are composed by more than one aminoacidic chain. Every polypeptidic chain these proteins are composed of is a subunit. The spatial arrangement, the structure, of the subunits in their entirety is called the quaternary structure of such proteins, also called protein complexes.
A protein characterized by a quaternary structure, composed of two, four, or more subunits will be called a dimer, a tetramer, and a multimer, respectively.
In some cases, the subunits are identical to each other, and in this case they can be called monomers, forming a homodimer, a homotetramer or a homomultimer. More often, the quaternary structure of a protein will be composed by subunits with different aminoacidic sequences, forming heterodimers, heterotetramers or heteromultimers. Examples of heteromultimers are histones, the protein complexes that keep the DNA of our genomes tightly packed in the cell nuclei, RNA polymerase, which transcribes messenger RNA from our genes, and hemoglobin, that carries oxygen in our bloodstream.
2.5. Protein folding#
We have repeatedly seen how protein function is linked to its structure, and how it’s possible to infer a protein function from its three-dimensional arrangement. But how do proteins assume the correct three-dimensional conformation starting from their primary sequence?
The driver of protein folding is escape from the solvent, a phenomenon that explains why hydrophobic patches (composed by hydrophobic amino acids) within a protein sequences tend to cluster together first, to form the hydrophobic core of the protein.
GOOD TO KNOW
The hydrophobic core amino acids are not necessarily close to each other in the primary sequence!
A notable exception to this principle are transmembrane proteins, for which the hydrophobic regions tend to be exposed to the outside environment – a special case where the outside environment is hydrophobic.
Copyright © 2018 Concord Consortium. The software is licensed under the MIT license.
2.6. Post-translational modifications#
Protein translation occurs in the cytoplasm thanks to the ribosomes. After translation, some proteins are subject to further processing by covalent enzymatic modification of specific residues in the Golgi apparatus. In some instances, these post-translational modifications (PTMs) are required for the protein function; others are involved in protein signaling, in cell cycle changes, or address the protein to a specific cellular destination.
NEED TO KNOW
Each modification relies on the presence on the target residue of a defined chemical group; therefore, not all PTMs can occur on each of the 20 amino acids.
There exist many types of PTMs, some common examples of which are phosphorylation, acetylation and glycosylation. Some PTMs have a more defined function in cell signaling, examples include SUMOylation and ubiquitination.
John F. Atkins and Ray Gesteland. The 22nd amino acid. Science, 296(5572):1409–1410, 2002. doi:10.1126/science.107333.